Description
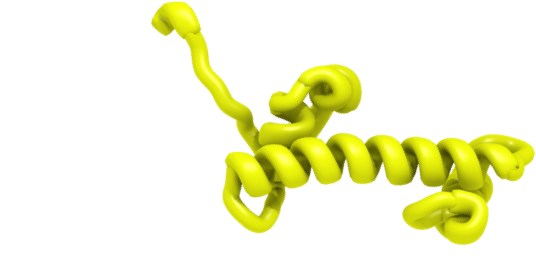
H2A is one of the core histones. H2A forms dimers with H2B via the "hand shake" motif. Two H2A-H2B dimers in turn associate with H3-H4 tetramer to form complete nucleosome core. Structure of H2A consists of histone fold domain extended by a short αC-helix and has both N- and C-terminal tails. αC-helix and C-terminal tail form "docking domain" that locks the H2A-H2B dimer onto the surface of H3-H4 tetramer.Histone Variants
37 / 8482
canonical H2A
Alternate names: ca H2A
Taxonomic span: Eukaryotes 2 / 1328 H2A.1
Alternate names: TH2A, TS H2A.1
Taxonomic span: Mammals 15 / 285 H2A.B
Alternate names: H2A.Bbd, H2A.Lap1(mouse)
Taxonomic span: Mammals 17 / 257 H2A.L
Alternate names: H2A.Lap2, H2A.Lap3, H2AL, H2AL1, H2AL2
Taxonomic span: Certain mammals 11 / 141 H2A.P
Alternate names: CXorf27, H2A.Lap4, HIP17, Huntingtin-interacting protein M, HYPM
Taxonomic span: Placentalia 9 / 2092 H2A.W
Alternate names: H2A with SPKK motifs
Taxonomic span: Plants 23 / 2487 H2A.X
Alternate names: member X
Taxonomic span: Eukaryotes except nematode 26 / 5492 H2A.Z
Alternate names: D2, H2A.V, H2A.Z, H2A.Z-1, H2A.Z-2, H2A.Zc, H2Av, H2AvD, Htz1p, hv1, member Z
Taxonomic span: Eukaryotes 10 / 2436 macroH2A
Alternate names: macroH2A1, macroH2A1.1, macroH2A1.2, macroH2A2, macroH2A2.1, macroH2A2.2, mH2A
Taxonomic span: Vertebrates(?)
Alternate names: ca H2A
Taxonomic span: Eukaryotes 2 / 1328 H2A.1
Alternate names: TH2A, TS H2A.1
Taxonomic span: Mammals 15 / 285 H2A.B
Alternate names: H2A.Bbd, H2A.Lap1(mouse)
Taxonomic span: Mammals 17 / 257 H2A.L
Alternate names: H2A.Lap2, H2A.Lap3, H2AL, H2AL1, H2AL2
Taxonomic span: Certain mammals 11 / 141 H2A.P
Alternate names: CXorf27, H2A.Lap4, HIP17, Huntingtin-interacting protein M, HYPM
Taxonomic span: Placentalia 9 / 2092 H2A.W
Alternate names: H2A with SPKK motifs
Taxonomic span: Plants 23 / 2487 H2A.X
Alternate names: member X
Taxonomic span: Eukaryotes except nematode 26 / 5492 H2A.Z
Alternate names: D2, H2A.V, H2A.Z, H2A.Z-1, H2A.Z-2, H2A.Zc, H2Av, H2AvD, Htz1p, hv1, member Z
Taxonomic span: Eukaryotes 10 / 2436 macroH2A
Alternate names: macroH2A1, macroH2A1.1, macroH2A1.2, macroH2A2, macroH2A2.1, macroH2A2.2, mH2A
Taxonomic span: Vertebrates(?)
Phylogenetic tree of variants (curated sequences)
Above, alignments of curated sequences from all H2A variants have been combined to construct the phylogenetic tree. Most of the variants cluster in separate clades. Click on the taxa name to learn more about its variant. A set of manually selected and validated histone sequences is listed in the table. Click on an entry in the table to update the annotated sequence preview: a variant will be compared with the canonical histone from the same species (if available).
Alternatively, tick mark the sequences and use toolbar to view MSA, export or add to basket. Use search or filters to find particular entries.
Keys: red - identical residues, blue - different residues (if more than one sequence). For feature legend see summary tab.
Sequence preview and annotation... LOADING
Note: variant classification might be ambigous between very similar variants. Classification scores against all variant models are available via advanced menu.
Features characteristic for a given histone type/variant are marked below the consensus sequence. For feature description see summary tabs of the corresponding variants pages.
Keys: red - 80% identical, blue - 50% identical columns. X-ambigous positions in consensus sequence.