Description
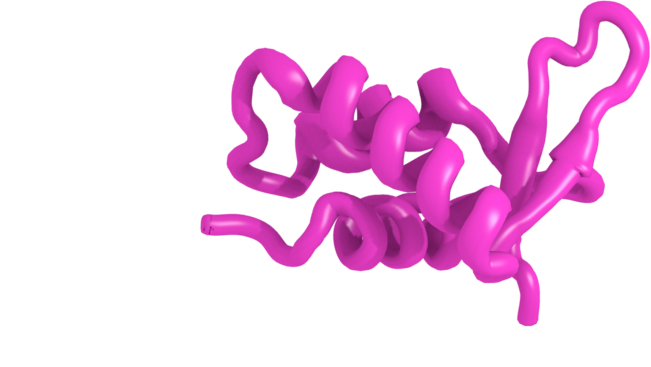
H1 is the linker histone. Associates with the nucleosome core and linker DNA near the DNA entry-exit points. The resulting particle is called chromatosome. H1-histone is lysine rich, has long disordered C-terminal tail and a short N-terminal tail. The globular domain has three helices and a wing, so-called "winged helix" motif.Histone Variants
18 / 10123
generic H1
Alternate names: gen H1
Taxonomic span: Eukaryotes 15 / 1344 H1.0
Alternate names: H1°, H1δ, H5, RI H1
Taxonomic span: Metazoa 6 / 278 H1.10
Alternate names: H1x
Taxonomic span: Vertebrates 2 / 494 OO H1.8
Alternate names: H1oo
Taxonomic span: Mammals 2 / 784 scH1
Alternate names: None
Taxonomic span: Saccharomyces(?) 8 / 753 TS H1.6
Alternate names: H1t
Taxonomic span: Mammals 2 / 227 TS H1.7
Alternate names: H1T2
Taxonomic span: Mammals 4 / 148 TS H1.9
Alternate names: Hils1
Taxonomic span: Mammals
Alternate names: gen H1
Taxonomic span: Eukaryotes 15 / 1344 H1.0
Alternate names: H1°, H1δ, H5, RI H1
Taxonomic span: Metazoa 6 / 278 H1.10
Alternate names: H1x
Taxonomic span: Vertebrates 2 / 494 OO H1.8
Alternate names: H1oo
Taxonomic span: Mammals 2 / 784 scH1
Alternate names: None
Taxonomic span: Saccharomyces(?) 8 / 753 TS H1.6
Alternate names: H1t
Taxonomic span: Mammals 2 / 227 TS H1.7
Alternate names: H1T2
Taxonomic span: Mammals 4 / 148 TS H1.9
Alternate names: Hils1
Taxonomic span: Mammals
Phylogenetic tree of variants (curated sequences)
Above, alignments of curated sequences from all H1 variants have been combined to construct the phylogenetic tree. Most of the variants cluster in separate clades. Click on the taxa name to learn more about its variant. A set of manually selected and validated histone sequences is listed in the table. Click on an entry in the table to update the annotated sequence preview: a variant will be compared with the canonical histone from the same species (if available).
Alternatively, tick mark the sequences and use toolbar to view MSA, export or add to basket. Use search or filters to find particular entries.
Keys: red - identical residues, blue - different residues (if more than one sequence). For feature legend see summary tab.
Sequence preview and annotation... LOADING
Note: variant classification might be ambigous between very similar variants. Classification scores against all variant models are available via advanced menu.
Features characteristic for a given histone type/variant are marked below the consensus sequence. For feature description see summary tabs of the corresponding variants pages.
Keys: red - 80% identical, blue - 50% identical columns. X-ambigous positions in consensus sequence.