Background information
Nucleosome structure
Nucleosomes are the basic units of chromatin compaction. Nucleosome core particle (NCP) wraps around 147 bp of DNA in ~1.7 left-handed super helical turns around an octamer of four types of core hisotnes (H3, H4, H2A and H2B — two copies of each) [1,2]. NCPs are further connected by linker DNA strands and a linker histone H1 further interacts with the NCPs and linker DNA to facilitate formation of higher order chromatin structures. Nucleosomes play key roles in epigenetic regulation of transcription, replication, DNA repair and cell reprogramming.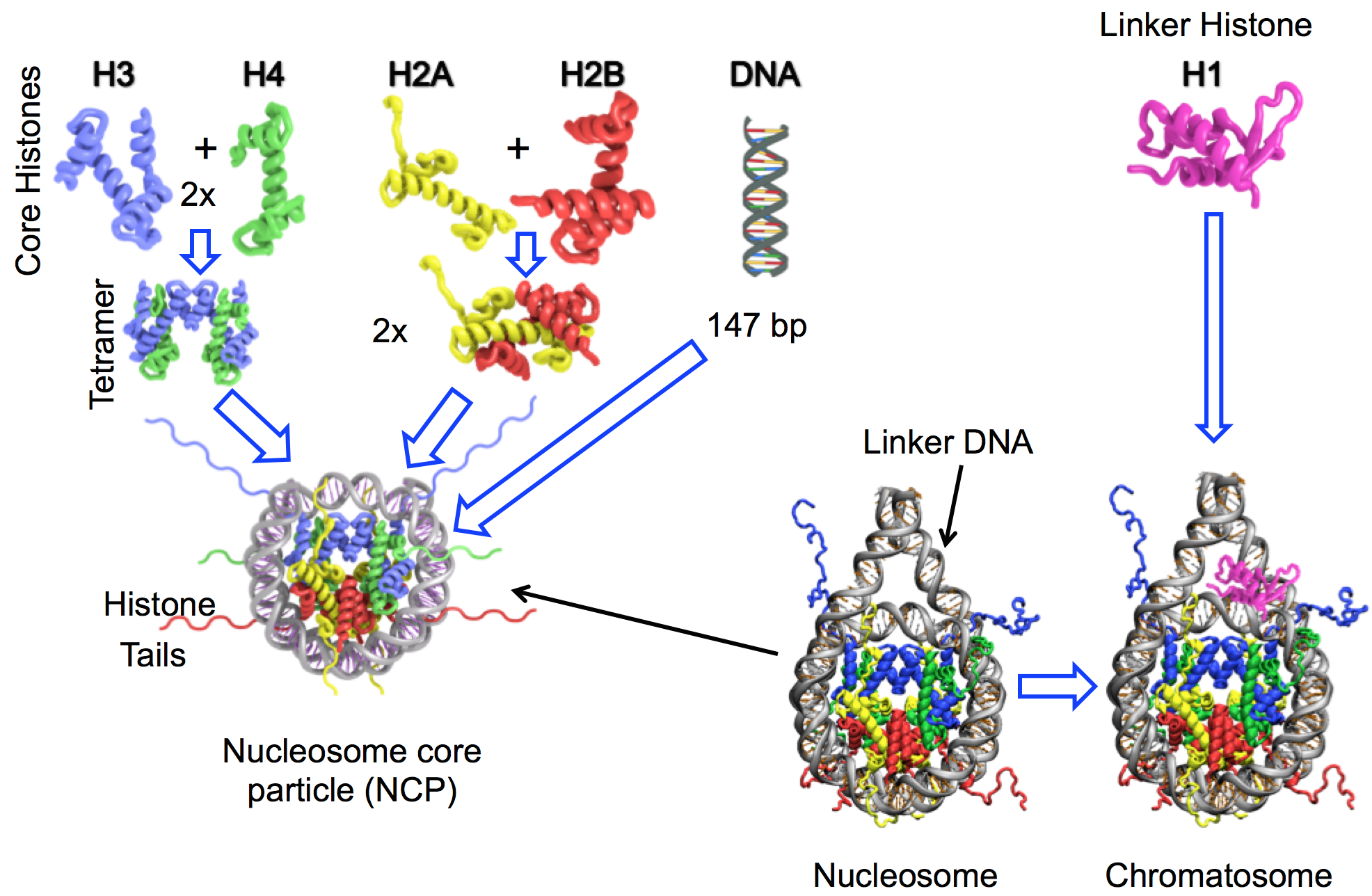
Nucleosome organization diagram (adapted from [3-4]).
Dynamic representation of the nucleosome with linker DNA [4].
Histone variants
Most eukaryotes have histone isoforms for some or all of the histone families (H2A, H2B, H3, H4, H1) [5]. Classification of histone isoforms is a daunting task. They are usually subdivided into ‘canonical’ replication-dependent histones that are expressed during the S-phase of cell cycle and replication-independent histone ‘variants’, constitutively expressed during cell cycle. In animals, genes encoding canonical histones are typically clustered along the chromosome, lack introns, and employ a specific type of regulation at the RNA level with a stem loop structure at the 3’ end instead of polyA tail. On the other hand, genes encoding histone variants are usually not clustered, have introns, and their mRNAs are regulated with polyA tails similar to the mRNAs of most genes. In plants, canonical histone genes lack introns, but are not clustered and the mRNAs are polyadenylated. Remarkably, more complex multicellular organisms typically have a higher number of histone variants providing a variety of different functions. Recent data are accumulating about the roles of diverse histone variants highlighting the functional links between variants and the delicate regulation of organism development.References
- Kornberg RD. "Chromatin structure: a repeating unit of histones and DNA." Science, 1974. PMID: 4825889
- Luger K, Mader AW, et al. "Crystal structure of the nucleosome core particle at 2.8 A resolution." Nature, 1997. PMID: 9305837
- Shaytan AK, Landsman D, et al. "Nucleosome adaptability conferred by sequence and structural variations in histone H2A-H2B dimers." Curr Opin Struct Biol, 2015. PMID: 25731851
- Shaytan AK, Armeev GA, et al. "Coupling between histone conformations and DNA geometry in nucleosomes on a microsecond timescale: atomistic insights into nucleosome functions." J Mol. Biol., 2015. PMID: 26699921
- Talbert PB, Ahmad K, et al. "A unified phylogeny-based nomenclature for histone variants." Epigenetics Chromatin, 2012. PMID: 22650316
Contact
Please, send your question and comments to histonedb-help@ncbi.nlm.nih.gov.
Nomenclature
This database of histone variant follows the new nomenclature introduced in ref. [1], which may be summarized in the following form:[prefix]TYPE.VARIANT
- TYPE = Histone type name (H2A, H2B, H3, H4, H1)
- VARIANT:
- Letter: used to denote a structurally distinct monophyletic clade of a histone family (exception is H2A.X).
- Number: used to denote a species-specific variant, not defined by phylogeny but clear orthology.
Example: H2A.Z
References
- Talbert PB, Ahmad K, et al. "A unified phylogeny-based nomenclature for histone variants." Epigenetics Chromatin, 2012. PMID: 22650316
Classification
The automatic classification algorithms employed in the current database is described below. More details available in ref.[1].Curated alignments of histone variants were used to train Hidden Markov Models, utilizing HMMER 3.1b2, to create one HMM for each variant. These models were used as a part of automatic extraction and annotation pipeline. Namely, all sequences from the nr protein database have been classified by the HMM models (variants and canonical) and were added into the HistoneDB 2.0 as “automatically extracted sequences”. We assigned a model with a maximum HMMER score to a given sequence. The score was required to exceed a certain threshold identified as follows. For any given variant model, we searched the curated sequence set and calculated HMMER scores for all curated sequences. We then varied the HMMER score threshold in order to distinguish variant sequences from all other sequences with 90% specificity. Specificity of retrieval was estimated based on the number of true positives (TPs; sequences correctly predicted by their native model) and false positives (FPs; incorrectly predicted sequences) found above each HMMER score cutoff. The specificity was calculated as TN/(FP+TN). Then a desired score cutoff value was obtained from the interpolated inverse curve of the score cutoffs plotted versus specificity. The efficacy of classification based on HMM models depends significantly on whether the respective histone variant model is sufficiently divergent and forms a separate clade on the phylogenetic tree. For certain variants, which emerged repeatedly in the course of evolution, the classification becomes difficult unless a characteristic signature is known. In the case of H2A.X, HMM search algorithm was supplemented by the pattern matching, since this variant is characterized by the presence of ‘SQ(E/D)Φ’ motif as described above. Any sequence that was classified as canonical H2A by HMMER-based algorithm was reclassified as H2A.X if it had this motif at its C-terminus. The described above classification algorithm can be also applied to classify any sequence of interest via "Analyze Your Sequence" tab.
References
- Draizen EJ, Shaytan AK, Marino-Ramirez L, Talbert PB, Landsman D, Panchenko AR. "HistoneDB 2.0 - with Variants: an integrated resource to explore histones and histone variants." Database. In Preparation.
How to cite
Please cite the database as:E.J. Draizen, A.K. Shaytan, L. Marino-Ramirez, P.B. Talbert, D. Landsman, A.R. Panchenko. "HistoneDB 2.0 - with Variants: an integrated resource to explore histones and histone variants." Database. In Preparation.